Finding the variants
that others miss.
Active Research Areas
___
My Variant Coach investigates rare, novel, and unresolved genetic variants in conditions that standard panels do not capture. Our research is grounded in published literature, phenotype-driven analysis, and direct submission to public scientific databases.
1 in 10
4–7
ClinVar
people are affected by a rare disease. Most have a genetic cause. Many remain undiagnosed for years.
years is the average time to a rare disease diagnosis. We work to shorten that journey.
Active submitter to the NIH-supported public archive of human genetic variants and disease.
Research Focus Areas
___
Three areas of active investigation
Each research area below represents an active focus at My Variant Coach, conditions where standard genetic reporting frequently returns negative or uncertain results despite the presence of clinically relevant variants.
01
Neuromuscular
NIPA1 & Atypical & Novel Variant Presentations
NIPA1 encodes a magnesium transporter critical for axonal integrity and corticospinal tract signaling. Known pathogenic variants in this gene cause SPG6, but the full disease spectrum of NIPA1, particularly for rare and novel variants outside the canonical mutation sites, remains poorly characterized.
This matters because patients with atypical NIPA1-related presentations frequently receive negative genetic reports or alternative diagnoses. The clinical picture may include episodic muscle fatigue, bulbar involvement, fluctuating EMG findings, and orofacial symptoms; features that fall outside traditional SPG6 descriptions.
Atypical presentations may include bulbar symptoms, jaw involvement, episodic weakness, and fluctuating EMG, features not typical of classic SPG6 but documented in complex NIPA1 cases.
Symptoms that may warrant NIPA1 investigation:
Possible symptoms and findings include: progressive proximal weakness, episodic muscle fatigue, bulbar involvement, jaw tremor, fluctuating EMG, neurogenic biopsy findings, intermittent exercise intolerance, and orofacial symptoms.
My Variant Coach investigates NIPA1 variants using phenotype alignment, splice prediction, segregation analysis where family data is available, and comprehensive literature review. Novel variants identified through this process may be candidates for ClinVar submission with client consent.
My Variant Coach is currently conducting observational crowdsourcing research to identify individuals with rare or novel NIPA1 variants who present without classic SPG6 features. This research was initiated following a de-identified clinical observation. If you or a family member carries a rare NIPA1 variant and does not have a confirmed diagnosis, we encourage you to reach out.
Episodic muscle weakness
Worsening with exercise
Jaw tremor
02
Mitochondrial Disease & Complex I–V Deficiency
MITOCHONDRIAL
03
X-linked & sex-chromosome variants
SEX LINKED
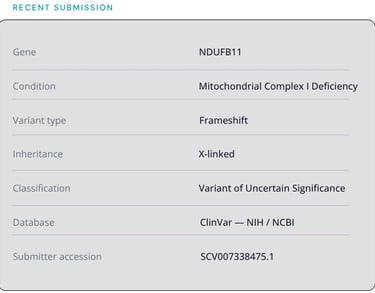
NDUFB11 encodes a structural subunit of mitochondrial Complex I, the largest component of the respiratory chain and the most common site of mitochondrial disease-causing variants. Located on the X chromosome, NDUFB11 follows an X-linked inheritance pattern, meaning expression and severity can differ significantly between males and females, and between generations.
The published phenotype for NDUFB11 centers on severe infant-onset presentations. However, the full disease spectrum of this gene remains poorly characterized. Childhood-onset presentations with milder or atypical features, including exercise intolerance, episodic fatigue, muscle weakness, and multi-system involvement without classic infant findings may represent a distinct phenotype that standard panels frequently miss or misclassify.
A 2024 Mayo Clinic study of 94 adults diagnosed with mitochondrial myopathy found a median of 11 years between symptom onset and diagnosis. Many had childhood onset but were not diagnosed until adulthood. This diagnostic gap, and the wide phenotypic spectrum documented, supports the importance of investigating mitochondrial variants even when presentation does not match classic descriptions.
Mitochondrial disease can look completely different from one family member to the next. One person may have severe fatigue, another may have muscle weakness, and another may have neurological symptoms. This variability is why it is so frequently missed.
Symptoms that may warrant NDUFB11 investigation:
Exercise intolerance
Progressive weakness
Fatigue and extreme exhaustion
Muscle pain
Peripheral Neuropathy
Neurological symptoms
Biological sex is not binary. Chromosomal patterns, hormonal biology, and genetic expression exist on a spectrum and medicine has not caught up with that reality.
Women are systematically underrepresented in genetic and rare disease research. Diagnostic criteria and symptom profiles have been built predominantly from male data, meaning women with the same condition are frequently misdiagnosed, dismissed, or told their condition doesn't affect their sex.
Intersex and DSD individuals, those whose chromosomal, hormonal, or anatomical sex development does not fit typical binary definitions are even further outside that research picture. Prevalence estimates vary enormously because rigorous population-level genomic research simply hasn't been done. We don't actually know how many people fall outside binary biological sex definitions. That absence of data is itself a medical problem.
This research area investigates how genetic conditions present across the full spectrum of biological sex, including in women, intersex individuals, and those with sex chromosome differences, and whether current diagnostic criteria, treatment protocols, and disease models adequately account for that variation.
Because they probably don't. And people deserve to be seen.
To be included in this study we will send an extra form for you to fill out with questions after we confirm your chromosome and variant findings. Certain labs do full coverage of chromosomal regions while others do not. Participation in this study may depend on which lab you were sequenced through.
OUR RESEARCH APPROACH
___
How we investigate complex genetic findings
Every investigation at My Variant Coach follows a structured research process designed to build a scientifically defensible, physician-ready picture of your genomic findings.
We begin with your phenotype which is your symptoms, your history, your family patterns. We use that to guide where in the genome we look and what we're looking for. We do not produce generic variant lists. We produce targeted, contextualized research.
All findings are classified conservatively, documented with published citations, and clearly framed as research for physician discussion, never as a diagnosis.
PHENOTYPE FIRST ANALYSIS
Your symptoms and clinical history guide variant prioritization. We build your phenotype before we look at variants.
SPLICE PREDICTION
We use computational splice prediction to evaluate whether intronic or synonymous variants may affect gene function in ways standard annotation misses.
Where family genomic data is available, we compare variants across family members to strengthen the evidence for candidate findings.
Every variant included in a report is supported by published research. We use only peer reviewed medical literature to build reports your doctor can evaluate.
SEGREGATION ANALYSIS
Literature SUPPORTED REPORTS
CLINVAR SUBMISSION
Novel variants meeting criteria are submitted to ClinVar with client consent, contributing findings to the global scientific record.
SCIENTIFIC CONTRIBUTIONS
___
Contributing to the public scientific record
When My Variant Coach identifies a novel variant with sufficient evidence to warrant broader scientific attention, we submit it to ClinVar, the NIH-supported public archive of human genetic variants and their relationship to disease.
This means that research performed through My Variant Coach has the potential to contribute to the body of knowledge that future patients, researchers, and clinicians draw from. Every submission is performed with full client consent and classified conservatively.
Novel variants are submitted as Variants of Uncertain Significance (VUS), the scientifically honest classification for a variant where evidence supports investigation but functional confirmation is pending.

Want to participate in any of our studies?
We're looking for three types of people: those who already carry a rare variant in one of these genes, those with symptoms that fit but no diagnosis yet, and those with a genetic test who received a negative result but fit these symptoms or diagnoses. If any of that describes you, we want to hear from you.